Append a new dataset¶
We have one dataset in storage and are about to receive a new dataset.
In this notebook, we’ll see how to manage the situation.
import lamindb as ln
import bionty as bt
import readfcs
bt.settings.organism = "human"
→ connected lamindb: testuser1/test-facs
ln.context.uid = "SmQmhrhigFPL0000"
ln.context.track()
→ notebook imports: bionty==0.49.1 lamindb==0.76.4 pytometry==0.1.5 readfcs==1.1.8 scanpy==1.10.2
→ created Transform('SmQmhrhigFPL0000') & created Run('2024-09-06 08:30:40.674919+00:00')
Ingest a new artifact¶
Access  ¶
¶
Let us validate and register another .fcs file from Oetjen18:
filepath = readfcs.datasets.Oetjen18_t1()
adata = readfcs.read(filepath)
adata
AnnData object with n_obs × n_vars = 241552 × 20
var: 'n', 'channel', 'marker', '$PnR', '$PnB', '$PnE', '$PnV', '$PnG'
uns: 'meta'
Transform: normalize  ¶
¶
import pytometry as pm
pm.pp.split_signal(adata, var_key="channel")
pm.pp.compensate(adata)
pm.tl.normalize_biExp(adata)
adata = adata[ # subset to rows that do not have nan values
adata.to_df().isna().sum(axis=1) == 0
]
adata.to_df().describe()
| CD95 | CD8 | CD27 | CXCR4 | CCR7 | LIVE/DEAD | CD4 | CD45RA | CD3 | CD49B | CD14/19 | CD69 | CD103 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| count | 241552.000000 | 241552.000000 | 241552.000000 | 241552.000000 | 241552.000000 | 241552.000000 | 241552.000000 | 241552.000000 | 241552.000000 | 241552.000000 | 241552.000000 | 241552.000000 | 241552.000000 |
| mean | 887.579860 | 1302.985717 | 1221.257257 | 877.533482 | 977.505533 | 1883.358298 | 556.687953 | 929.493316 | 941.166747 | 966.012244 | 1210.769935 | 741.523184 | 1003.064857 |
| std | 573.549695 | 827.850302 | 672.851319 | 411.966073 | 584.217139 | 932.113729 | 480.875917 | 795.550133 | 658.984751 | 456.437094 | 694.622980 | 473.287558 | 642.728024 |
| min | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 |
| 25% | 462.757715 | 493.413744 | 605.463427 | 588.047798 | 495.437303 | 1063.670965 | 240.623098 | 404.087640 | 477.932659 | 592.294399 | 575.401173 | 380.247262 | 475.108131 |
| 50% | 774.350833 | 1207.624048 | 1110.367681 | 782.939692 | 782.981430 | 1951.855099 | 484.355203 | 557.904360 | 655.909639 | 800.280049 | 1124.574275 | 705.802991 | 775.101973 |
| 75% | 1327.792103 | 2036.849496 | 1721.730010 | 1070.479036 | 1453.929567 | 2623.975657 | 729.754419 | 1345.771633 | 1218.445208 | 1347.042403 | 1742.288464 | 1069.175380 | 1420.744291 |
| max | 4053.903716 | 4065.495666 | 4095.351322 | 4025.827267 | 3999.075551 | 4096.000000 | 4088.719985 | 3961.255364 | 3940.061146 | 4089.445928 | 3982.769373 | 3810.774988 | 4023.968008 |
Validate cell markers  ¶
¶
Let’s see how many markers validate:
validated = bt.CellMarker.validate(adata.var.index)
! 9 terms (69.20%) are not validated for name: CD95, CXCR4, CCR7, LIVE/DEAD, CD4, CD49B, CD14/19, CD69, CD103
Let’s standardize and re-validate:
adata.var.index = bt.CellMarker.standardize(adata.var.index)
validated = bt.CellMarker.validate(adata.var.index)
! 7 terms (53.80%) are not validated for name: CD95, CXCR4, LIVE/DEAD, CD49B, CD14/19, CD69, CD103
/tmp/ipykernel_3171/92294437.py:1: ImplicitModificationWarning: Trying to modify index of attribute `.var` of view, initializing view as actual.
adata.var.index = bt.CellMarker.standardize(adata.var.index)
Next, register non-validated markers from Bionty:
records = bt.CellMarker.from_values(adata.var.index[~validated])
ln.save(records)
! did not create CellMarker records for 2 non-validated names: 'CD14/19', 'LIVE/DEAD'
Manually create 1 marker:
bt.CellMarker(name="CD14/19").save()
CellMarker(uid='3ZFziy5ims8J', name='CD14/19', created_by_id=1, run_id=2, organism_id=1, updated_at='2024-09-06 08:30:45 UTC')
Move metadata to obs:
validated = bt.CellMarker.validate(adata.var.index)
adata.obs = adata[:, ~validated].to_df()
adata = adata[:, validated].copy()
! 1 term (7.70%) is not validated for name: LIVE/DEAD
Now all markers pass validation:
validated = bt.CellMarker.validate(adata.var.index)
assert all(validated)
Register  ¶
¶
curate = ln.Curator.from_anndata(adata, var_index=bt.CellMarker.name, categoricals={})
curate.validate()
• 1 non-validated categories are not saved in Feature.name: ['LIVE/DEAD']!
→ to lookup categories, use lookup().columns
→ to save, run add_new_from_columns
✓ var_index is validated against CellMarker.name
True
curate.add_validated_from_var_index()
artifact = curate.save_artifact(description="Oetjen18_t1")
• path content will be copied to default storage upon `save()` with key `None` ('.lamindb/cWmcWgk8zKjnt0Sj0000.h5ad')
✓ storing artifact 'cWmcWgk8zKjnt0Sj0000' at '/home/runner/work/lamin-usecases/lamin-usecases/docs/test-facs/.lamindb/cWmcWgk8zKjnt0Sj0000.h5ad'
• parsing feature names of X stored in slot 'var'
✓ 12 terms (100.00%) are validated for name
✓ linked: FeatureSet(uid='w1u4DxZh1VNafLgwfa1i', n=12, dtype='float', registry='bionty.CellMarker', hash='r6ixHrg-eAYhYva4So6xwQ', created_by_id=1, run_id=2)
• parsing feature names of slot 'obs'
! 1 term (100.00%) is not validated for name: LIVE/DEAD
! skip linking features to artifact in slot 'obs'
✓ saved 1 feature set for slot: 'var'
Annotate with more labels:
efs = bt.ExperimentalFactor.lookup()
organism = bt.Organism.lookup()
artifact.labels.add(efs.fluorescence_activated_cell_sorting)
artifact.labels.add(organism.human)
artifact.describe()
Artifact(uid='cWmcWgk8zKjnt0Sj0000', is_latest=True, description='Oetjen18_t1', suffix='.h5ad', type='dataset', size=46506448, hash='WbPHGIMM_5GT68rC8ZydHA', n_observations=241552, _hash_type='md5', _accessor='AnnData', visibility=1, _key_is_virtual=True, updated_at='2024-09-06 08:30:45 UTC')
Provenance
.created_by = 'testuser1'
.storage = '/home/runner/work/lamin-usecases/lamin-usecases/docs/test-facs'
.transform = 'Append a new dataset'
.run = '2024-09-06 08:30:40 UTC'
Labels
.organisms = 'human'
.experimental_factors = 'fluorescence-activated cell sorting'
Feature sets
'var' = 'Cd4', 'CD8', 'CD95', 'CD49B', 'CD69', 'CD103', 'CD14/19', 'CD3', 'CD27', 'CXCR4', 'Ccr7', 'CD45RA'
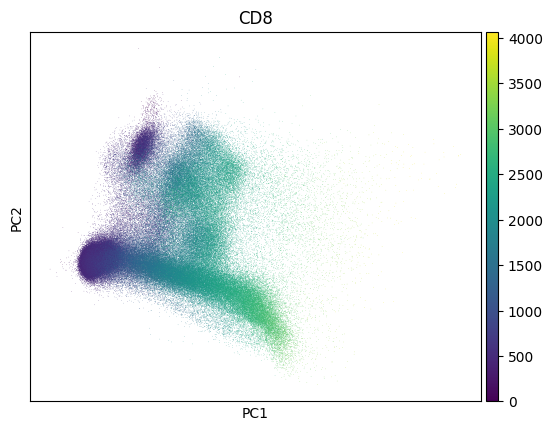
Inspect a PCA fo QC - this collection looks much like noise:
import scanpy as sc
markers = bt.CellMarker.lookup()
sc.pp.pca(adata)
sc.pl.pca(adata, color=markers.cd8.name)
Create a new version of the collection by appending a artifact¶
Query the old version:
collection_v1 = ln.Collection.get(name="My versioned cytometry collection")
collection_v2 = ln.Collection(
[artifact, collection_v1.ordered_artifacts[0]], is_new_version_of=collection_v1, version="2"
)
collection_v2.describe()
! `is_new_version_of` will be removed soon, please use `revises`
• adding collection ids [1] as inputs for run 2, adding parent transform 1
• adding artifact ids [1] as inputs for run 2, adding parent transform 1
Collection(uid='jw8pJy486fV81M2y0001', version='2', is_latest=True, name='My versioned cytometry collection', hash='aIyjTZDm9LEyi4udLlQ-FA', visibility=1)
Provenance
.created_by = 'testuser1'
.transform = 'Append a new dataset'
.run = '2024-09-06 08:30:40 UTC'
Feature sets
'var' = 'CD57', 'Cd19', 'Cd4', 'CD8', 'Igd', 'CD85j', 'CD11c', 'CD16', 'CD3', 'CD38', 'CD27', 'CD11B', 'Cd14', 'Ccr6', 'CD94', 'CD86', 'CXCR5', 'CXCR3', 'Ccr7', 'CD45RA'
'obs' = 'Time', 'Cell_length', 'Dead', '(Ba138)Dd', 'Bead'
collection_v2.save()
✓ saved 1 feature set for slot: 'var'
Collection(uid='jw8pJy486fV81M2y0001', version='2', is_latest=True, name='My versioned cytometry collection', hash='aIyjTZDm9LEyi4udLlQ-FA', visibility=1, created_by_id=1, transform_id=2, run_id=2, updated_at='2024-09-06 08:30:46 UTC')